基因克隆实验
==最后修改时间:2023年5月7日==
1 克隆基因
(1)准备物品
ddH
2O,一对引物
(确保引物TM值在55度左右(加臂前),且加上载体所需用的长臂序列)
1. PYES2载体臂:(用于酵母异源表达)Amp
F:cttggtaccgagctcggatcc;
R:tacatgatgcggccctctaga,
2. PBI121载体长臂:(可用于亚细胞定位,但需要(仅此和非gateway法根毛转化)去终止子,仅仅在R链,逆序读终止子)Kan
F:gagaacacgggggactctaga;
R:ccatggtacccccggggatccg
3. PDONR_RNAi(也可过表达)入门载体载体长臂: Zeo
F:GGGGACAAGTTTGTACAAAAAAGCAGGCTTC
R:GGGGACCACTTTGTACAAGAAAGCTGGGTC
4. Pgadt7载体载体长臂:AD(加转录因子)NdeI EcorI(单杂,双杂都用) Amp
F:gtaccagattacgctcatatg
R:atgcccacccgggtggaattc
5. PGBKT7i载体长臂:BD(加基因)NdeI EcorI(双杂用,双杂=AD+BD) Kan
F:tcagaggaggacctgcatatg
R:tcgacggatccccgggaattc
6. pAbAi载体长臂:SmaI SalI(单杂用,单杂=AD+pAbAi),AD-MYB111,PABAI-WD40.
F:aattcgagctcggtacccggg
R:agcacatgcctcgaggtcgac
7. pGreenII-0800载体长臂:(双荧光素,验证转录因子与启动子结合活性)HindIII BamHI
F:gtcgacggtatcgataagctt
R:cgctctagaactagtggatcc
1 | 8.双分子荧光互补载体长臂:(验证两基因或转录因子间的互作)非无缝克隆 |
pSAT1-cCFP-C(pE3242):
F:GGAATTCG(EcoRI)flycut
R:CGGGATCC(BamHI)
pSAT1-nVenu-C(pE3228):
F:CGAGCTCGC(SacI)1.5XT
R:ACGCGTCGACG(Sali)+BSA
注意:双分子荧光互补实验中==不仅需要酶切载体,还需要酶切胶回收产物。==
1 | 9.PBI121-gus载体长臂:(在启动子中,验证基因表达位置,需要染色)HindIII BamHI,pbi121-gus酶切后需要跑胶后切胶 |
F:GACCATGATTACGCCAAGCTT
R:GGACTGACCACCCGGGGATCC
cDNA,红BUFFER,蓝高保真酶,绿
琼脂糖,1*TAE,胶回收试剂盒(天根或生工),纯化试剂盒
工具:八联管,冰盒,冰砖,离心管,记号笔
仪器:PCR机,凝胶成像仪器
(2)实验步骤
①高保真酶体系(八连管中每1管加入25μL,每基因2-3重复)
| 高保真酶体系配置 total:25μL | |||
|---|---|---|---|
| 1 | cDNA | 1μL | 自己准备的东西 |
| 2 | 引物1 | 1μL | |
| 3 | 引物2 | 1μL | |
| 4 | ddH |
8.5μL | |
| 5 | 红 | 12.5μL | buffer(2 x phanta Max Buffer) |
| 6 | 蓝 | 0.5μL | 高保真酶,最后加,不能在常温过久(Max super Fidelity) |
| 7 | 绿 | 0.5μL | DNTP |
②按照上述体系将各组分加入八连管中,进行短时离心,9000X。
③将八连管放入PCR机,程序DZ/NWC,改退火温度为自己的温度,这里为55度,1000bp延伸时间为 1min,进行扩增,退火后的一步时间为1000bp为1min,600bp可为50s。
同时制备胶,若有10个以内样品,用半幅胶,为0.75g琼脂糖加1 * TAE50ml,后放入微波炉按照30s,取出混匀,20s,取出混匀,20s,取出混匀完,至全透明步骤进行。若10个以上,为全幅胶,为1.5g琼脂糖加1 * TAE100ml。在倒入电泳槽前加入==核酸染料1滴,2μL==,混匀。
④扩增结束后,在(八连管中每1管)样品中加入Loading Buffer==2==μL,使用枪头混均匀。
⑤将混好的样分别点入琼脂糖凝胶,在最开始的位置点入marker4-6μL(最开始之位置为胶孔对着我们的方位时,最右一个胶孔)。
⑥进行电泳20min,135v。若发现条带已经在中间或更远位置,拿出胶放入凝胶成像仪,按照302nm灯光成像,查看是否有条带分离,远离点样区的是二聚体,即为引物等,靠近点样区的是我们要回收的cDNA。
⑦将同一基因的胶块切下,应该注意需要尽快,不可长期在302nm下光照,否则会导致DNA结构破坏,而且要使切下的胶块尽可能的少琼脂部分。
⑧==跑出胶后宜立马胶回收。==
⑨面向我们从右往左点样。
PS:大孔胶可加样25μL,小孔胶可加样4-10μL。
2 胶回收(电泳结束后宜立即执行)
①将切下的胶放入15μL离心管,之后按照胶回收试剂盒要求进行即可。若暂时不进行以下操作,可放入4度冰箱。
②需要注意的是我们基因的碱基数,还有配置胶的浓度,这些和加入b2溶液多少有关。
③另外还要注意的是,空柱离心后,务必要换新的的收集管,即为15μL离心管,最后一步我们回收的产物是离心管内的物质,而非柱子上的物质。若暂时不进行下一步操作放入-20度冰箱。(CDNA,DNA,质粒,RNA皆为-20℃保存)
3 酶切(PDONR不需要此步骤)
①加液体系(不同质粒(质粒浓度之不同)之酶切体系不同)
(以下步骤皆在冰砖上进行)
| 酶切体系(pYES2) | |||
|---|---|---|---|
| 1 | PYSE2 | 4μL | 合计20μL |
| 2 | BamHI | 1μL | |
| 3 | XBaI | 1μL | |
| 4 | 10Xk | 1μL | |
| 5 | ddH2O | 13μL |
| 酶切体系(pET32a) | |||
|---|---|---|---|
| 1 | PYSE2 | 4μL | 合计20μL |
| 2 | BamHI | 1μL | |
| 3 | Sac I | 1μL | |
| 4 | 10Xk | 1μL | |
| 5 | ddH2O | 13μL |
| 酶切体系(pBI121-gus) | 浓度为40ng/μL | ||
|---|---|---|---|
| 1 | pBI121-gus | 16μL | 合计20μL |
| 2 | BamHI | 1μL | |
| 3 | Hind III | 1μL | |
| 4 | 10Xk | 2μL |
| 酶切体系(pBI121) | 浓度为40ng/μL | ||
|---|---|---|---|
| 1 | pBI121 | ==37μL== | 合计50μL |
| 2 | ==BamHI== | ==5μL== | |
| ==3== | ==Hind III== | ==5μL== | |
| ==4== | ==10Xk== | ==3μL== |
| 酶切体系(pSAT1-cCFP/nVenu) | 浓度为40ng/μL | ||
|---|---|---|---|
| 1 | ==pSAT1-cCFP/nVenu== | ==16μL== | ==合计20μL== |
| ==2== | ==BamHI== | ==1μL== | |
| ==3== | ==Hind III== | ==1μL== | |
| 4 | ==10Xk== | ==2μL== |
PS:酶切体系中需用载体1μg,若载体浓度为50ng/μL,则此载体1ng为20μL,因体系之最大为20μL,且各酶占用1+1+2=4μL,故该载体需要加入16μL。
②放入PCR仪37度1hour(后可4℃保存,当日使用,可放-20℃,半年内使用)
③==酶切产物需要用纯化试剂盒纯化==。
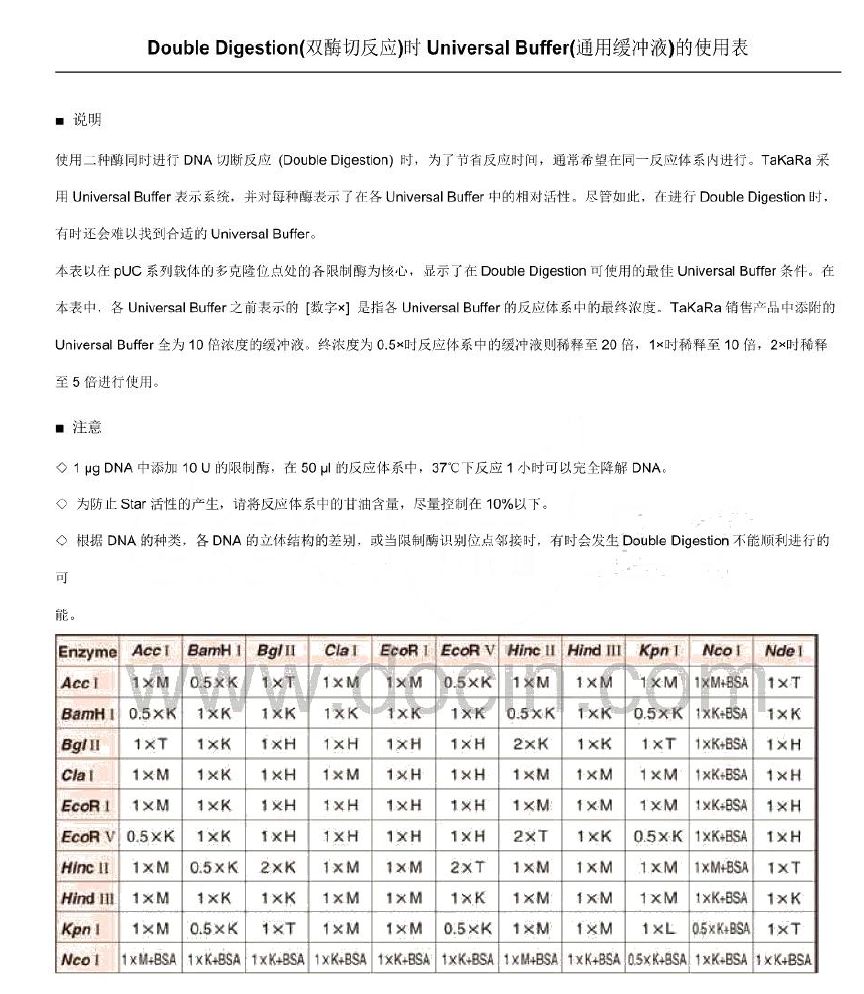
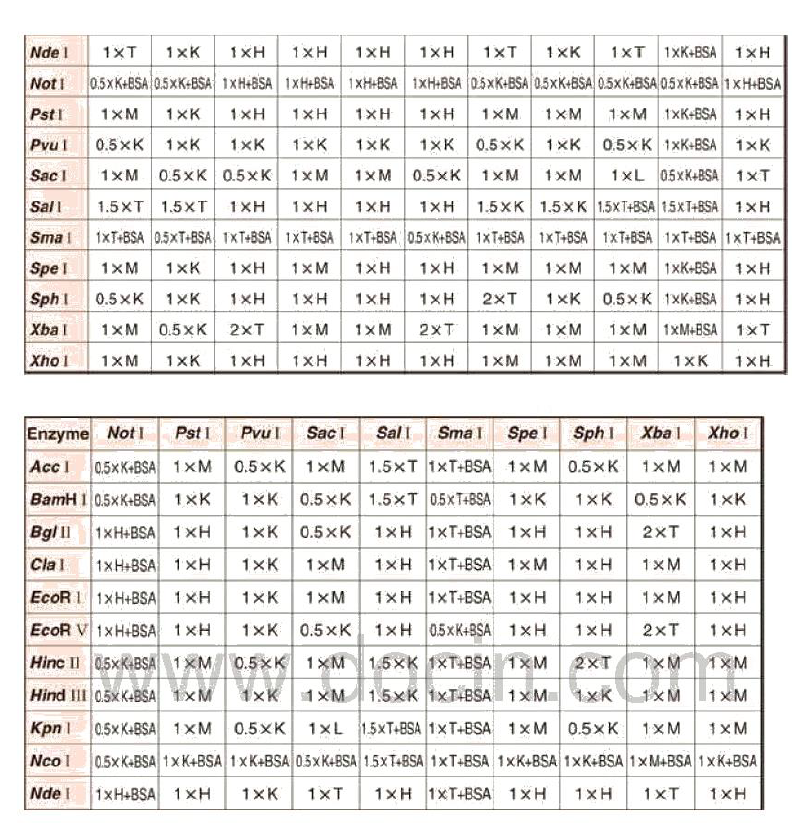
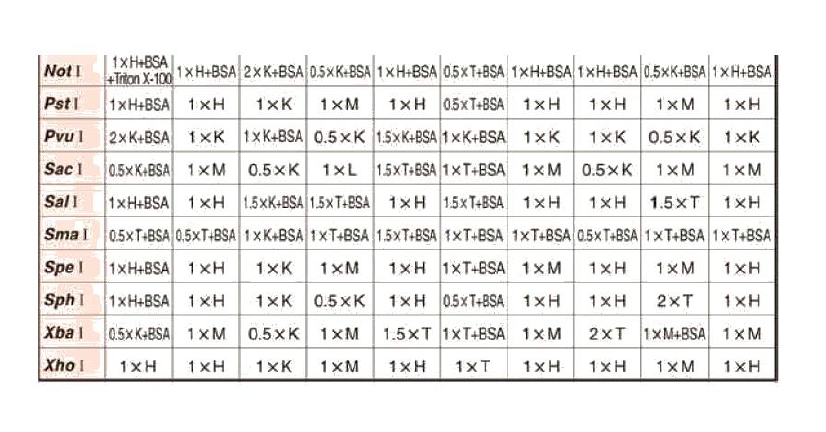
4 转入大肠杆菌(连接)
①加液体系
| 连接体系 | |||
|---|---|---|---|
| 1 | PCR回收产物 | 1μL | 合计5μL |
| 2 | 酶切产物 | 2.5μL | |
| 3 | Esnase | 0.5μL | |
| 4 | CE buffer | 1μL |
==Esnase 和 CE buffer有的试剂盒中合为一个试剂==
②放入PCR仪37度30min(后可4℃保存,当日使用)
③加入连接产物5μL 至33 (或50)μL DH5aα(又名trans 5α) ,放入冰盒内静置 30 min,使用金属浴42度45s,后放入冰盒固定位置4-5min,加入500μL LB液体。(若为其他大肠杆菌如BL21(DE3),(尝用pET32a时使用)则改trans5α为其他)
④摇菌1h37度,将制备好的菌液放入4度冰箱保存。
⑤配置LB固体培养基,应当提前进行,需要高温灭菌,同时灭菌培养皿,其配方为:
| LB固体液体培养基 1L体系 | |||
|---|---|---|---|
| 1 | 水 | 1L | 液体 |
| 2 | 蛋白胨 | 10g | |
| 3 | 酵母 | 5g | |
| 4 | NaCl | 10g | |
| 5 | 琼脂粉 | 15g | 固体额外加入 |
PS:Zeo之LB固体培养基配法为上述Nacl用量之1/2,其余用量皆同。
⑥若加入抗生素,灭菌后1L内加入1ml安定Amp。(特别的,Zeo为1L内加入333μL,其余皆同Amp用量)
从⑥步骤开始都要在灭菌操作台,手套和工具都要喷酒精进行。
⑦将LB固体培养基倒入培养皿,倒至合理位置即可。
⑧待固体培养基凝固后,使用涂菌棒,把菌液均匀涂抹至培养基。(涂200μL菌液)
⑨使用封口膜封口。
⑩放入菌的培养箱内。
5 检测阳性菌
①加入到八连管内的物质体系:
| 阳性检测体系 | |||
|---|---|---|---|
| 1 | 蓝(2xSan Tag PCR MIX) | 5μL | 合计10μL乘9倍为90微升 使用引物类似的离心管,加8个就弄10倍 |
| 2 | ddH |
3μL | |
| 3 | 引物1 | 1μL | |
| 4 | 引物2 | 1μL |
②瞬时离心,9000X。
③拿出前面培养的菌(应当为24H后,且有肉眼可见的菌斑),在新的amp的LB固体培养基底部用记号笔划线,最好为一个板子上分为两半,一半划线16条。
④将原来的培养基内的斑块先用枪头挑出,在新的培养基记号笔划线处用该枪头划线,之后用该枪头在八连管内顺时针方向旋转将菌混匀至八连管内。
⑤结束后,将原来的和新的培养皿都进行封口膜封口,放入菌的培养箱。
⑥将混匀好的八连管放入PCR仪内,进行扩增。同时制备胶。
⑦结束后,在八连管内加入 ,把marker点至第一个孔(只点marker),其余的样点样至胶板上,进行135v,20min的电泳,进行凝胶成像。
⑧若呈现阳性,可以找出对应的菌落,混入离心管,先加入500μLLB培养液,再加入1μL Amp,最后混入离心管(北京奥科,填写测序登记表装袋)送去公司进行测序。

